 PROTOCOL PROCEDURES
PROTOCOL PROCEDURES
How long is the washout period?
We consider the day -7 (V2) as the last day of the screening period, while the washout period is 6 days long, from day -6 to day -1 included (Study Manual – Section: Wash-out period- page 11). Therefore, patients who are non-naive to chelation therapy will suspend their ongoing chelator from day -6 to day -1 (with the last chelator administration no later than 24:00 on day -7). If the last dose of chelator is expected to be administered to the patient after midnight of day -7 then the patient should not take it, in order to comply with the protocol procedure which recommends the drug suspension from the first minute after midnight.
Should treatment-naïve patients go through the wash-out period?
Chelation-naïve patients are not expected to go through the washout period and can perform V2 the same day of V3 Also patients who have suspended chelation therapy more than 6 days prior to V2 do not need to go through the washout period, and can be randomized after verification of all inclusion/ exclusion criteria.
When will the clinical site receive the PK kit?
As soon as you complete the Section- Drug Management of V14 in the eCRF, an alert will be sent to the Sponsor Pharmacokinetic contact (
[email protected]) for the attribution of your site sampling time and material supply. Sampling scheme and complete kit will be provided to your site.
If my fax is not available or out of order, how should I notify a Serious Adverse Notification to the Pharmacovigilance Service?
After the SAE form is duly filled in, validated on the Pharmacovigilance section of the e-CRF, downloaded as pdf, printed and signed, it must be sent to the Safety Contact to start the processing of the Serious Adverse Event within 24 hours of learning of its occurrence.
In case the fax is not available, the SAE form can be forwarded also via email (email address
[email protected]).
The dispatch of SAE forms via email is deemed as official as the forwarding via fax also for the notification of all confirmed Neutropenia episodes (two consecutive measurements), which, irrespective of their severity or seriousness, will follow the procedure for SAE notification and therefore must be reported by the Investigator to the Sponsor within 24 hours of learning of their occurrence.
What should I do if the icon of a battery appears on the display of the temperature data logger I use to monitor the storage temperature of the IMP?
The flashing battery icon stands for “low battery”. Once connected to the pc, a window warning that the device would not be able to record the temperatures anymore will appear. Therefore I should promptly replace the battery following these few steps:
– connect the device to the pc through the USB wire;
– download and archive the file RECORD produced;
– disconnect the logger from the pc;
– unscrew the battery slot and replace it;
– restart the temperature data logger and continuing monitoring the temperatures of the IMP.
What is the maximum and the minimum interval to perform Visits from 1 (screening) to 3 (randomization)?
The maximum interval allowed to perform V1 to V3 is 28 days. We recommend to shorten this time when possible in order to have enough time to make up delay or re-program unattended visits.
For not naive patients (who require washout) the V2 (washout period) has to be performed at day -7 from the V3 (V2 and V3 have to fall on the same day of two consecutive weeks, e.g. V2 on Monday and V3 on the consecutive Monday). The washout period cannot be shortened at all.
For naive patients (who do not need washout) V2 and V3 can be coincident and the minimum interval between V1 and V2/V3 can be even of 1 day if laboratory tests are available and checked before performing the next visit.
When and how do I perform randomization?
- After having verified with the patient that wash out period is properly completed; and
- After having checked laboratory tests required for the fulfilment of inclusion/exclusion criteria (Hb, Neutrophil count, platelet count and serum creatinine assessed at visit 2 or alternatively at -9 or -8 from V3); and
- After having performed clinical examination and blood withdrawal required at V3.
You have to log on to the e-CRF to access the randomization form of V3. Once completed the randomization confirmation screen, the randomization option will appear among V3 activities. For more detailed information, please refer to the Study Manual vers. 2.0 (page 9) or ask CRA or TMT for support.
When is cardiac MRI T2* required according to the protocol?
Cardiac MRI T2* has to be performed in patients aged 10 years or more on the day of the first MRI (required at V3 +/- 20 days) and without need for sedation.
BLOOD SAMPLE HANDLING
What does the protocol statement: “serum creatinine must be assessed in duplicate (for the assessment of inclusion/exclusion criteria only)” mean?
At Visit 1, all lab tests, as per protocol, have to be performed. At Visit 2 (-7) or at -9/-8, complete blood count and serum creatinine have to be retested, in order to check the inclusion/exclusion criteria. So the assessment “in duplicate” means once at V1 and once at V2.
What do I do if a blood sample for local determination is lost?
As soon as the patient come back to the hospital, e.g. for a transfusion, the missed analysis should be repeated. Laboratory tests necessary for dose adjustment or safety monitoring (e.g. neutrophil count, renal function tests, liver enzymes, local serum ferritin), as required from the protocol, should be collected as soon as possible in order to guarantee patient’s safety. Please contact your CRA, who will advise you how to record the delayed blood tests, specifying the actual date of sampling
What do I do if a blood sample for central ferritin determination is lost?
As soon as the patient come back to the hospital, e.g. for a transfusion, the missed analysis should be repeated, avoiding any additional needle puncture. Please contact your CRA, who will advise you how to record the delayed blood test, specifying the actual date of sampling.
What does the protocol statement: “all tests on hepatitis serology will be carried out on the same sample as collected for blood haematology/biochemistry” mean?
The blood sample for hepatitis serology will be collected during the same withdrawal for Haematology/Biochemistry, but in a different test tube. If you miss the sampling for hepatitis serology at V1 or V15, you should plan a further assessment as soon as the patient has a blood sample, in order to avoid any additional needle puncture. Please contact your CRA, who will advise you how to record in eCRF the delayed blood tests, specifying the actual date of sampling.
IMP HANDLING
Is it possible to prefill the Exjade labels?
Not before the patient’s randomization.
How many label logs for DFP and DFX should be used?
One label log for each visit of each patient.
Can I dispense single blisters of Exjade to give to the patient the exact number of required tablets?
No, you cannot. All Exjade tablets must always be dispensed in their blisters, and the blisters must never be separated by the external packaging. This is an essential element of compliance with the ICH GMP. Please, pay great attention in organizing the visits combining both the requirements of the study protocol for the dates and assuring the dispensation of entire packages of Exjade (blisters + external box).
How do I dispense deferiprone between two consecutive visits?
The amount of Deferiprone to be dispensed at each visit/dispensation day must always cover the exact number of days expected between one visit/dispensation day and the following. The actual day of both visits/dispensation day must always be included during the calculation of the total volume to be dispensed. At each visit/dispensation day, all the drug received in the previous visit must be returned by the patient. I can then dispense new DFP bottles, keeping into account again the three daily doses of both the visit/dispensation-days. It is important to remember that, for the evaluation of patient’s compliance in terms of discrepancies between the actual remaining volume and the expected volume to be returned, the drug volumes consumed during the visit/dispensation-days will not be included in the calculation.
What should I do if a patient vomits after the dose intake?
If vomiting occurs because of a gastroenteritis or fever or a flu, the drug should not be administered. If patient has vomiting or spits because he/she does not want to take the drug, the dose can be repeated within 30 minutes after the first administration.
If vomiting occurs after 30 minutes, the dose should not be repeated. However a method to avoid vomiting should be devised (e.g. administration of the whole dose in little amounts -a single dose of 6 ml could be administrated in 3 separate doses of 2 ml within few minutes- or offering a sip of water after the drug ingestion).
Are Exjade tablets divisible?
No, they are not. Total daily dose of Exjade must be rounded to the nearest whole tablet size and must be not less than 20 mg/kg body weight.
Can the Investigator re-dispense an already allocated bottle of deferiprone?
Absolutely no. When a patient randomized to deferiprone comes back to a new visit, he/she must return all the used, unused and partially used bottles to the Investigator. The Investigator must then to:
- make sure of the patient compliance measuring the exact residual volume using the graduated 250 ml cylinder provided by the Sponsor;
- maintain record of the exact residual volume into the appropriate section of the eCRF;
- pour the volume again into the bottles;
- re-seal and withdraw all the bottles from the patients (empty or partially empty bottles);
- store all the returned bottles far from the other medicinal products for clinical use.
The stored bottles must be clearly identified as “Unusable Investigational Medicinal Products – DO NOT TOUCH” until the Clinical Monitor has verified the delivered, used and recovered quantities of product and, if applicable, once any discrepancies have been investigated and satisfactorily explained and the reconciliation has been accepted.
What if the patient didn’t return the IMPs?
The CRA has to:
1. report an asterisk near the treatment number in the Drug Dispensing and Accountability Log, write below the table: “The patient didn’t return the bottle”, then date and sign it,
2. collect the deviation to the protocol as stated in the SD.060 “Standard Operating Procedures for handling deviations and violations”.
The PI has to:
1. make a statement, signed by the PI, declaring that some IMPs didn’t return, specifying the batch number, the expiration date, the dispensation visit and the reason.
PATIENT ISSUES
Can I include patients under combined chelation therapy in the study?
As detailed in the study protocol, only patients that are in monotherapy just before their inclusion in the study can be enrolled.
Can a patient with high ALT values caused by muscular dystrophy be enrolled in the DEEP-2 study?
No, because the study treatment may further alter ALT values. Moreover, ALT level > 5 x ULN at screening is one of the exclusion criteria.
How do I define “contraindication” to DFO for a patient naïve to the chelation therapy, in order to include him/her in the study?
A contraindication to DFO, in addition to the medical contraindications, can be also based on the inability of the patient to achieve adequate adherence to parental treatment regimen. The reason for contraindication must be recorded in the medical chart.
Can other medications be administered to the patient during the study?
If the patient is in treatment, even chronically, with a drug that is not included among the prohibited medications (as described in the Protocol 3.0) and such medication does not preclude enrollment in the study, this treatment should not be interrupted during the washout period. If the drug is included in the prohibited medications and the treatment needs to be continued by the patient, he/she shall not be included in the study.
Subjects should abstain from taking any non-prescription medication, vitamins, herbal and dietary supplements within 7 days (or 14 days if the drug is a potential enzyme inducer) or 5 half-lives (whichever is longer) prior to the first dose of study drug (Day 0) until completion of the follow-up visit (V16, month 13). Vitamin supplementations should be prescribed just for verified deficiency.
In the case of an antibiotic treatment, it is advisable to wait until the end of the antibiotic cycle before starting the washout.
ADVERSE EVENT MANAGEMENT AND IMP DOSE ADJUSTMENTS
If an adverse event occurs during the treatment period, what should I do regarding the study treatment?
DFP and DFX can be scaled down for the safety reasons as detailed in the study protocol and summarized in the Study Procedure Manual- Section IMPs Dose Adjustment- Pages 16- 20.
How should any Serious Adverse Event (SAE) occurring during the trial be reported?
Any Adverse Event occurred between Vn and Vn+1, irrespective of the seriousness, should be always recorded in the Adverse Event section of Vn+1. When the event is deemed serious (Serious Adverse Event), “YES” must be selected in the last column of the form.
In case of seriousness, after the AE validation, an alert to remember the compilation of the SAE form in the Pharmacovigilance section (left sidebar of the e-CRF) will appear.
The SAE form must be completed in English. The duly filled-in SAE form must be then validated, downloaded as pdf, printed and sent via fax or email to the Safety Contact in DEEP-2 Safety/Efficacy Trial.
Instructions for management and rapid notification of Neutropenia events.
All confirmed neutropenia, defined as ANC < 1500/mm3, irrespective of their severity or seriousness, will follow the procedure for SAE notification and must be reported by the Investigator to the Sponsor within 24 hours of learning of their occurrence. “Confirmed” means that the ANC has been found to be within the specified range on 2 consecutive measurements. If both consecutive counts are below 1.5 x 109/L but are not within the same range, a third count is required to determine the severity category. Neutropenia is considered to be resolved when ANC is ≥1.5 x 109/L on 2 consecutive counts. A not confirmed neutropenia has not to be notified as SAE but as an AE in the specific section of e-CRF.
When a patient who experienced mild neutropenia can restart the IMP drug?
According to the emended version of the study protocol (3.0), when a mild neutropenia occurs, the IMP drug can be restarted at the last dose after two consecutive neutrophil counts are within the normal ranges and after consulting the Trial Management Team and the Coordinating Investigator respectively at the following addresses:
Maddalena Casale –
[email protected]
Aurelio Maggio –
[email protected]
IMP drug must be definitively interrupted when a moderate or severe neutropenia, confirmed in two consecutive measurements, occurs.
Which parameters should I evaluate in case of an early termination?
According to protocol V.3.0., in case of early termination visit will be evaluated: Haematology/ biochemistry, as reported in the protocol, centralized ferritin and CHQ. Please remember to collect all the partially used, unused, empty DFP bottles/DFX boxes and store them at your centre. You cannot redispense Exjade tablets used during the study after the early termination visit.
For those patients entering the trial with very high levels of ferritin, should I increase the dose of the drug if ferritin values remain stably high but do not increase further than the 20% margin as specified in the study protocol?
The observation period must be of 3 months. If, after this period, the ferritin values remain stably high (>1500 ng/ml without downtrend over a period of 3 months), therapy can proceed with a dose increase, as detailed in the Study Procedure Manual- Section: Dose adjustments for ferritin levels- page 16.
On which ferritin value can I perform dose adjustments?
I will evaluate whether to either confirm current dosing or scale up the drug dose, for efficacy reasons, on the base of local serum ferritin values. The study drug can be scaled up in case of persistent serum ferritin value > 1500 ng/ml without downtrend over a period of 3 months or increase of serum ferritin level > 20% compared to the previous determination.
If serum ferritin value is ≤500 ng/ml, the study drug has to be reduced of 50% of the last dose and the Coordinating Investigator, Prof. Aurelio Maggio ([email protected]) has to be contacted. Dose adjustment for ferritin levels are reported in the Study Procedure Manual- Section: Dose adjustments for ferritin levels- page 16.
If the patient by the time of inclusion in the study has scheduled surgery or any procedure requiring hospitalisation to be performed during the course of the trial, how can this be reported?
You do have to report the scheduled surgery or procedure in the patient’s medical chart prior or contextually to study inclusion. Once the surgery or procedure has occurred, it will need to be registered in the Healthcare Resources Section of the e-CRF. If decided prior or contextually to inclusion, the scheduled surgery or procedure will NOT have to be reported as SAE. Only if any event’s complication occurs, this event will be an AE/SAE depending on the seriousness.
If a patient experiences an adverse event which requires temporary chelation interruption, is the Investigator allowed to prescribe a different iron chelator in order to let the patient continue chelation therapy?
No, it is not allowed. Chelation drug assigned during randomization cannot be changed at all if the patient is still ongoing. Differently, this conduct would correspond to a severe violation. Investigators are strictly recommended to discuss with Study Coordinator and the Trial Management Team prior to withdrawing a patient from the study.
Neutropenia is defined as any ANC < 1500/mm3 confirmed in two consecutive measurements and reported as SAE. What is the time-frame to repeat the neutrophil assessment for confirmation?
In case of an abnormal neutrophil count (neutrophil value < 1500/mm3), a second assessment have to be performed within 48 hours from the first one. In case of moderate neutropenia (neutrophil count 500- 1000/mm3) or agranulocytosis (neutrophil < 500/mm3), it is recommended to repeat neutrophil count on the same day or within 24 hours from the previous assessment and interrupt IMPs even before confirmation.
What is the management of neutropenia?
The handling and follow up of the patient will depend on the severity of the neutropenia (mild, moderate,
severe/agranulocytosis) as follows.
Mild Neutropenia
– treatment must be interrupted and patient’s ANC must be monitored every 1-3 days, or more frequently, until resolution of the event, defined as two consecutive ANC ≥ 1500/mm3.
– if the patient has a mild neutropenia and develops signs of infection (fever), therapy must be interrupted immediately and ANC must be obtained and monitored every 1-2 days, or more frequently, until resolution of the event.
– provide protective isolation; treat the patient as per clinical need and per local protocol (antibiotic therapy, admission to hospital if clinically indicated). If possible, hemocolture, throat swab, viral studies (CMV, parvovirus, hepatitis A/B/C), serum ALT, BUN, creatinine will be collected.
– therapy with DFP or DFX and continuation of trial can be restarted once all symptoms have been resolved and when it is deemed safe by the Investigator, after having consulted the Coordinating Investigator, in accordance with Table 4, Section 6.3 “Dose Adjustment”.
Moderate Neutropenia
– treatment must be interrupted and the patient must be withdrawn from the study and monitored until
resolution of the event;
– provide protective isolation; if clinically indicated, admit patient to hospital, obtain regular vital signs
and treat the patient as per clinical need and per local protocol (antibiotic therapy and/or granulocyte
colony stimulating factor).
– the patient will be examined the same day, if possible, collecting drug history and physical examination;
– if possible, hemocolture, viral studies (CMV, parvovirus, hepatitis A/B/C), serum ALT, BUN, creatinine
will be collected.
Severe Neutropenia/Agranulocytos
– treatment must be interrupted immediately and the patient must be withdrawn from the study and monitored until resolution of the event;
– provide protective isolation; if clinically indicated, admit patient to hospital, obtain regular vital signs and administer appropriate therapy such as antibiotic therapy and granulocyte colony stimulating factor, beginning the same day that the event is identified;
– the patient will be examined the same day, if possible, collecting drug history and physical examination;
– if possible, hemocolture, viral studies (CMV, parvovirus, hepatitis A/B/C), serum ALT, BUN, creatinine will be collected.
– the patient will be monitored daily or more frequently until two successive ANCs are ≥ 1500/mm3.
e-CRF MANAGEMENT
When will the V3 screenshot appear in the e-CRF?
After V2 validation. V2 will appear after the validation of the “Informed consent and demographic data” in V1.
When do I have to enter visit data in the e-CRF?
For any patient, the e-CRF has to be completed and validated within 7 working days from the visit date. Furthermore, all forms of any ongoing patient have to be completed before the scheduled MOV.
When do I have to fill in the renal function in e-CRF?
Renal function (assessed as serum creatinine and creatinine clearance) must be determined:
• at each scheduled study visit (V4 to V15) for each patient;
• weekly during the first month after initiation;
• weekly after therapy’s modification. For example if at visit X (e.g. V5) you modify the therapy, you have to monitor the renal function weekly and fill it in the e-CRF at the next visit (in this case at V6).
When do I have to fill in the Healthcare Resources form in the e-CRF?
This form has to be completed only in case of use of resources (visits, tests or procedures) outside the study protocol. This means that every time your patient needs any additional clinical intervention, not already planned according to the study, you have to report it in this form.
What if the PI or his/her delegate wants to fill in the Drug Management section of the e-CRF,but one or more treatment numbers are missing on the dropdown menu?
PI has to:
• contact the CRA and explain the issue describing in details the missing treatment number/s and the Visit number of the patient;
CRA has to:
• communicate via email to the Data Manager ([email protected]) the issue, who will provide with the required support.
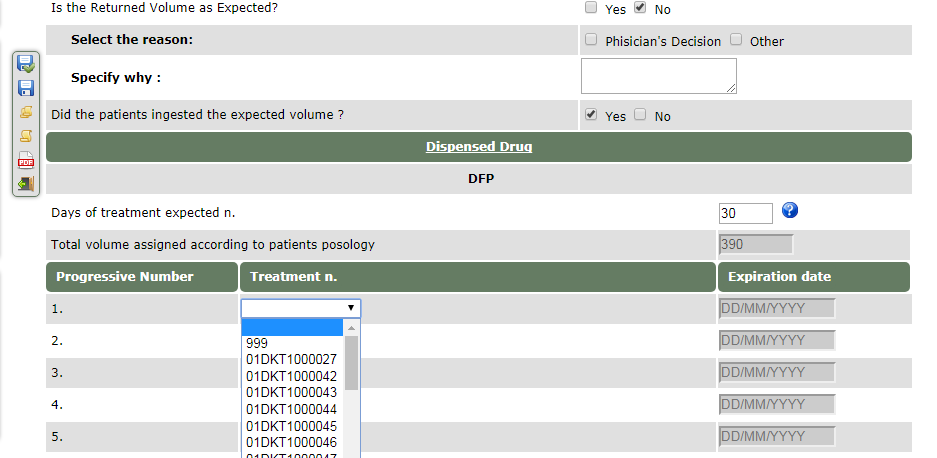
What if does not appear the correct treatment number in the drop down menu of the field “returned drug” or “dispensed drug” of the drug management section on the e-CRF?
A fake number as 999 can be chosen by the Investigators as shown below:
What if I cannot validate a form and the data seems correctly entered?
In case you are not able to validate a form even though all data have been correctly entered,
please contact the Data Manager to the email
[email protected]; you will be helped to
validate the form.

